Contact us
Need more information? Your local key account manager will be in contact shortly.
This page is intended for UK healthcare professionals and other relevant decision makers only. If you are a member of the public, please click here.
This portal is funded and owned by Novartis Pharmaceuticals UK Ltd and includes content approved by Novartis.
Adverse events reporting information can be found in the footer of this page.

Prescribing information (external link)
KESIMPTA is indicated for the treatment of adult patients with relapsing forms of multiple sclerosis (RMS) with active disease defined by clinical or imaging features.1
The most important and frequently reported adverse reactions are upper respiratory tract infections (39.4%), systemic injection-related reactions (20.6%), injection-site reactions (10.9%) and urinary tract infections (11.9%).1
For full safety information, please refer to the KESIMPTA Summary of Product Characteristics (SmPC).1
Click on the buttons below to explore the analyses from the open-label ALITHIOS extension study.
Data from exploratory analyses are descriptive and no confirmatory clinical conclusions can be drawn.
KESIMPTA met the primary endpoint in ASCLEPIOS I/II, with up to 58% reduction in ARR vs teriflunomide (ASCLEPIOS I: 51% [0.11 vs 0.22]; RR: 0.49 [95% CI: 0.37–0.65]; ASCLEPIOS II: 58% [0.10 vs 0.25]; RR: 0.42 [95% CI: 0.31–0.56]; both p<0.001).1,3
KESIMPTA has demonstrated efficacy in ASCLEPIOS I and II (2-year core studies), two Phase III randomised, double-blind, double-dummy, active comparator-controlled, parallel-group, multicentre pivotal trials, comparing a range of outcomes to active comparator teriflunomide in 1882 patients with RMS, following patients for up to 6 years (including the 4-year extension phase, ALITHIOS study).1–3
ALITHIOS is an ongoing, open-label, single-arm, umbrella-extension, Phase IIIb study assessing the risk–benefit profile of KESIMPTA (20 mg subcutaneously every 4 weeks) and its tolerability in patients with RMS.4
Adapted from Hauser SL, et al. 2022.4
ASCLEPIOS I and II primary endpoint: annualised relapse rate (ARR) was defined as the number of confirmed relapses of multiple sclerosis (MS) per year, according to prespecified criteria.3
ASCLEPIOS I and II secondary endpoints: disability worsening confirmed (CDW) at 3 months and 6 months, disability improvement confirmed (CDI) at 6 months, the number of gadolinium-enhancing (Gd+) lesions per T1-weighted magnetic resonance imaging (MRI) scan, the annualised rate of new or enlarging lesions on T2-weighted MRI, serum neurofilament light chain levels (NfL) at Month 3, and brain volume change (BVC).3
KESIMPTA met the primary endpoint in ASCLEPIOS I/II, with up to 58% reduction in ARR vs teriflunomide (ASCLEPIOS I: 51% [0.11 vs 0.22]; RR: 0.49 [95% CI: 0.37–0.65]; ASCLEPIOS II: 58% [0.10 vs 0.25]; RR: 0.42 [95% CI: 0.31–0.56]; both p<0.001).1,3
Use the arrows below to navigate to the 7-year recently-diagnosed (≤3 years) treatment-naïve (RDTN) data.
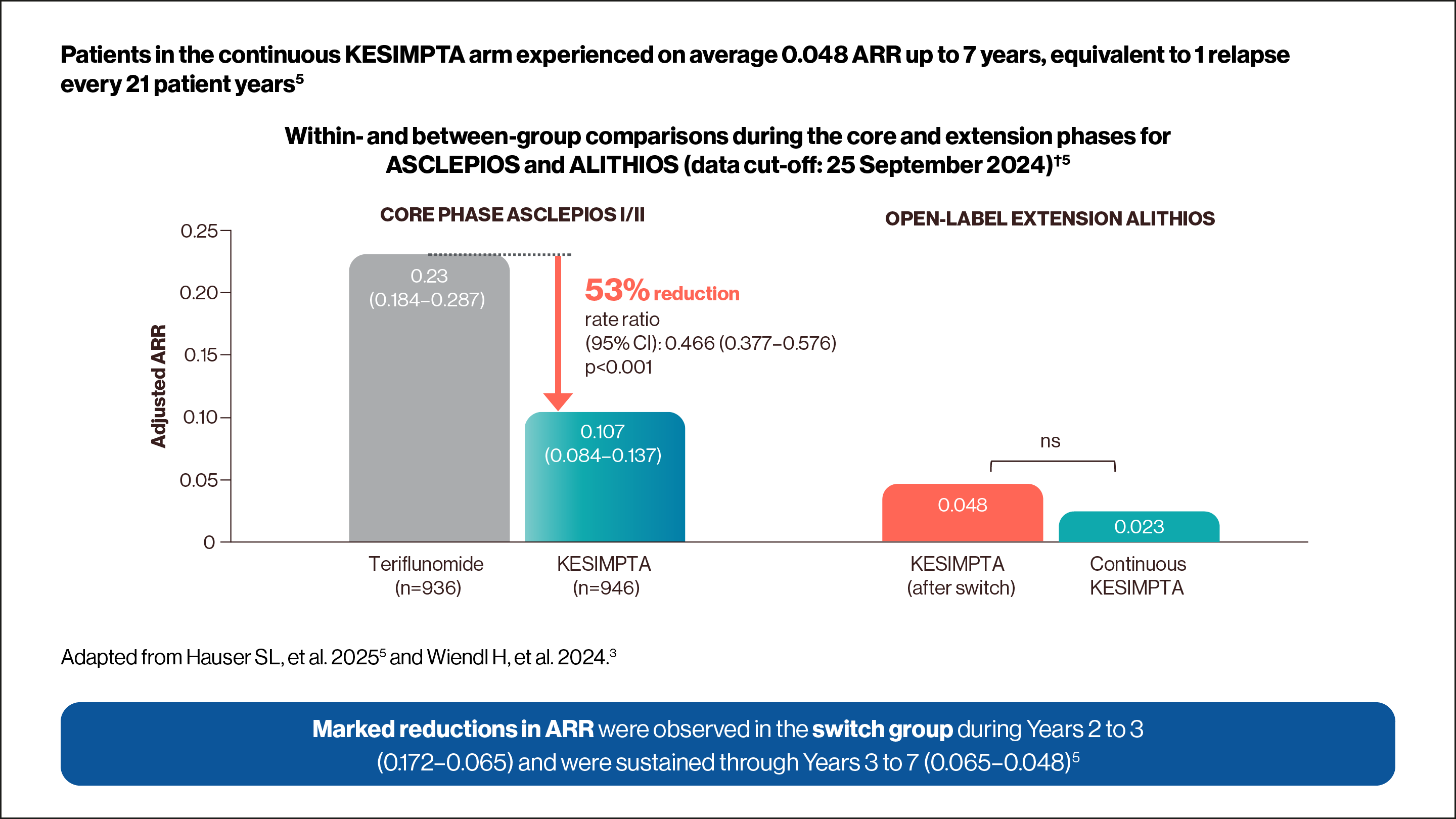
*ARR was defined as the number of confirmed MS relapses per year, according to prespecified criteria. Confirmed relapses are those accompanied by a clinically relevant change in expanded disability status scale (EDSS).2
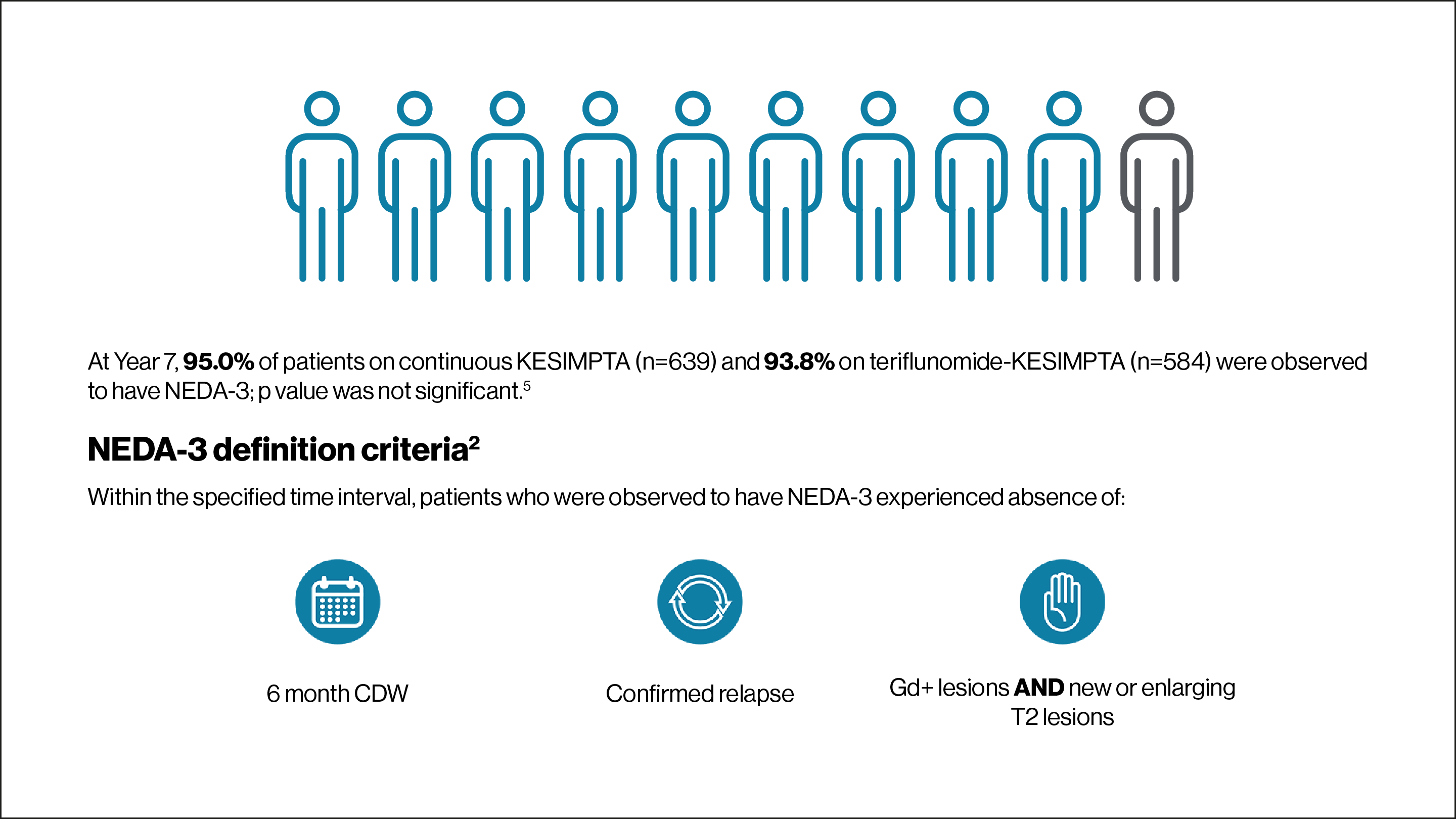
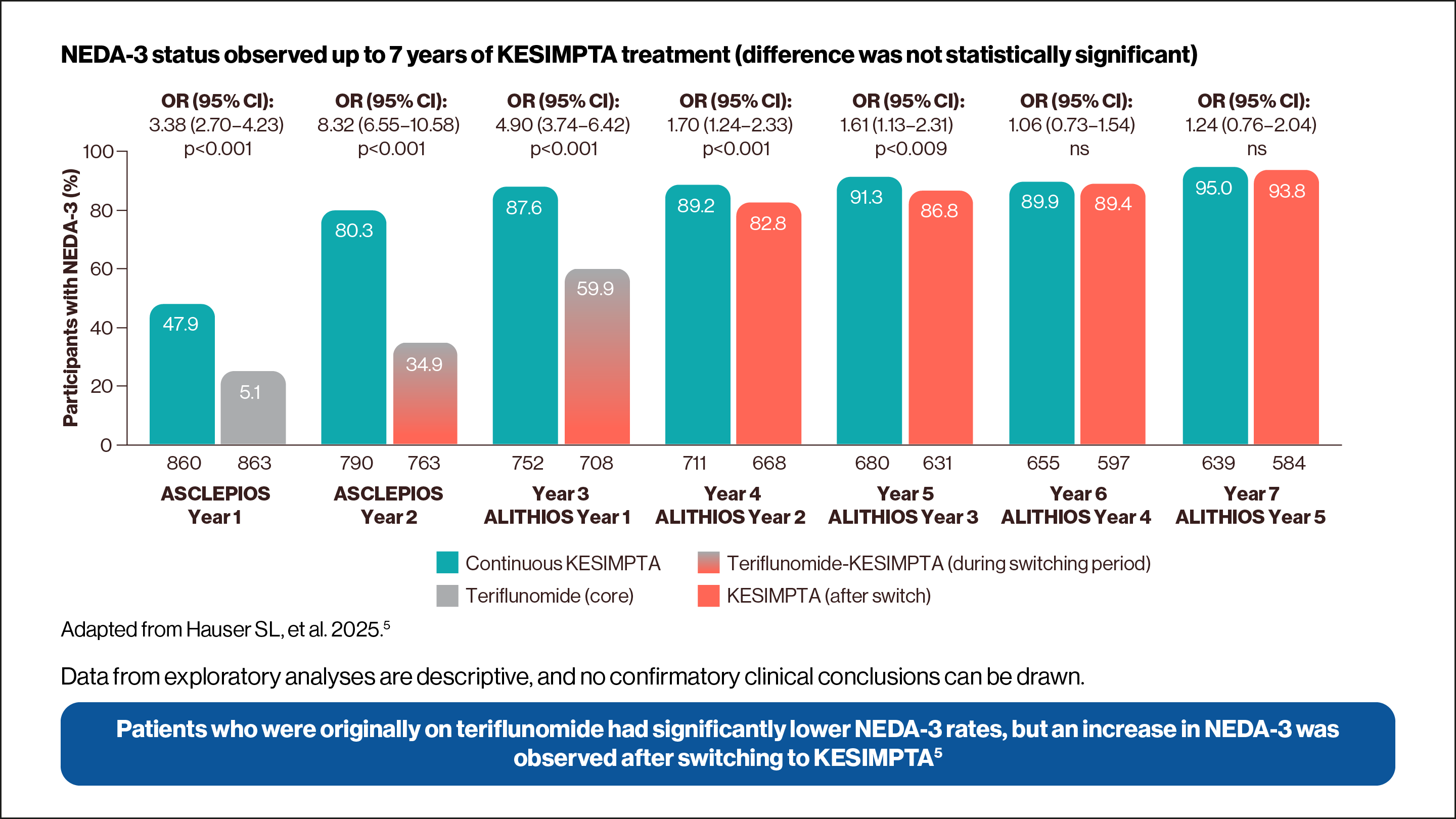
NEDA-3 was determined in a post-hoc analysis of pooled ASCLEPIOS trials.2,5
Data from exploratory analysis is descriptive, and no confirmatory clinical conclusions can be drawn.
Use the arrows below to navigate to the 7-year NEDA-3 and RDTN subgroup data.
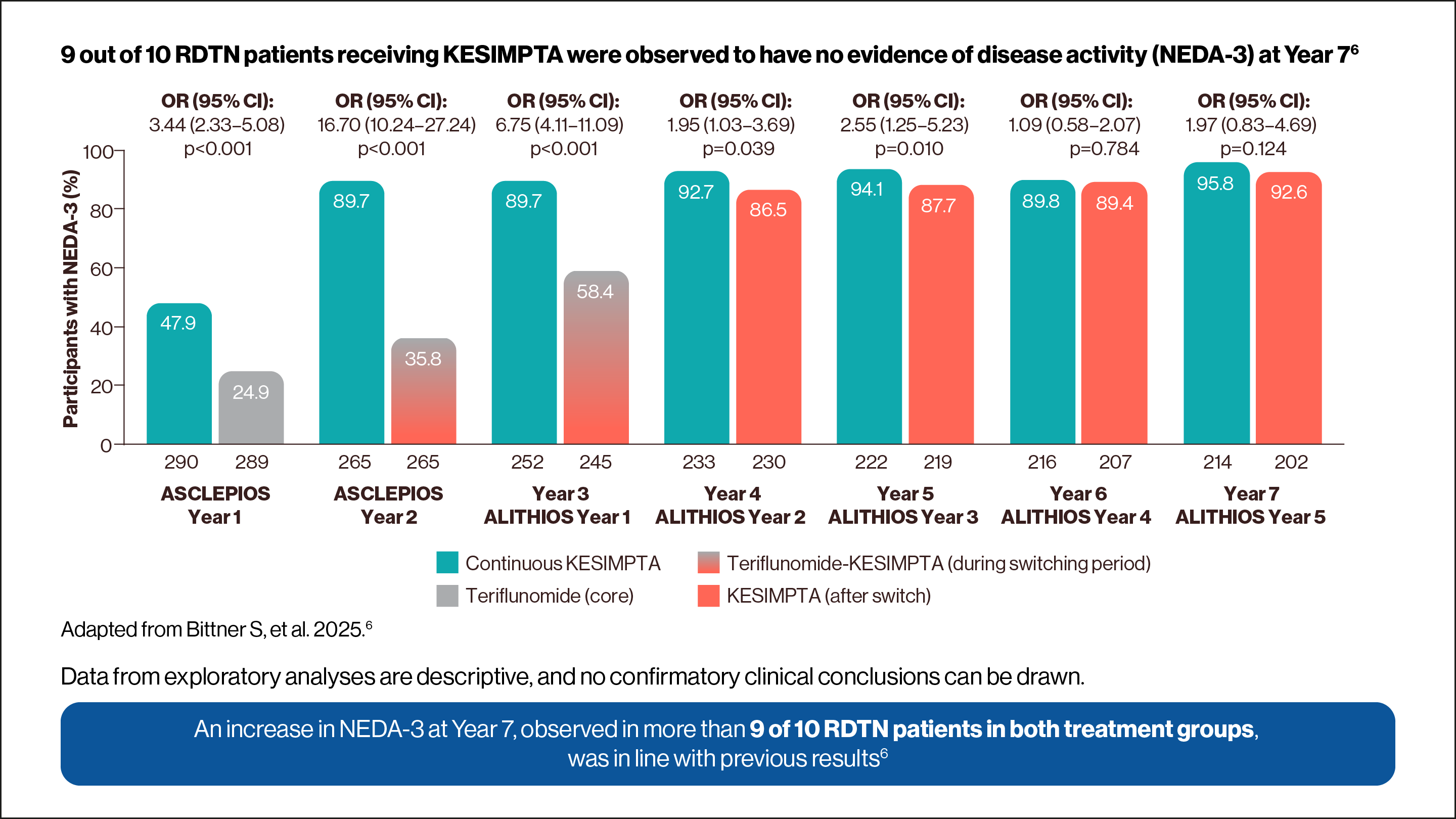
Adapted from Wiendl H, et al. 2024.2
Up to Year 7: An almost complete suppression of Gd+ T1 lesion activity (95.6% observed reduction for KESIMPTA vs teriflunomide in ASCLEPIOS I/II) was observed to be maintained from Years 1 to 7 in the continuous KESIMPTA group and from Years 3 to 7 in the switch group5 |
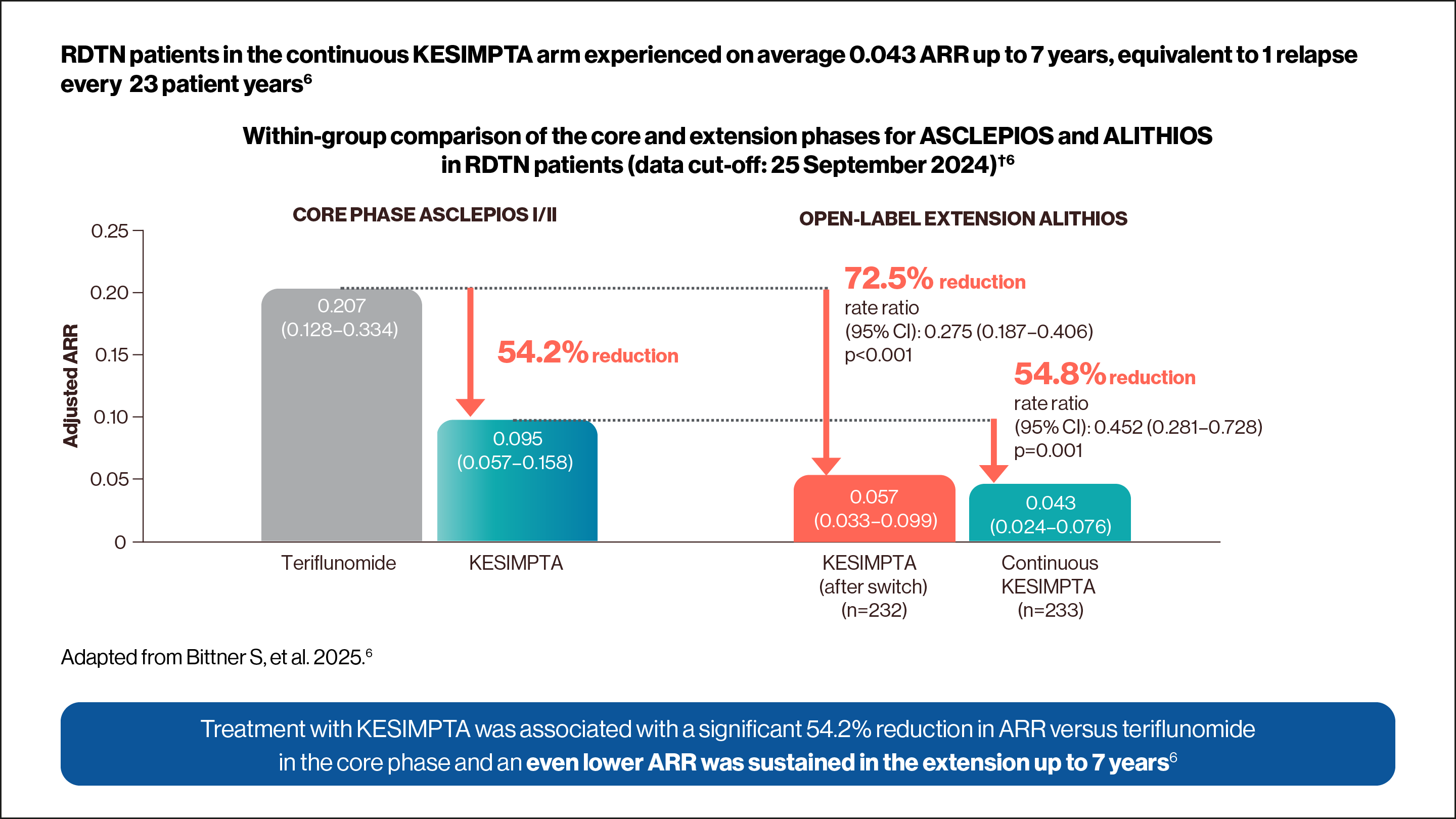
Within-group comparisons during the core and extension phases for ASCLEPIOS and ALITHIOS in recently-diagnosed treatment-naïve (RDTN) patients‡2
Adapted from Weindl H, et al. 2024.2
Use the arrows below to navigate to the RDTN subgroup data.
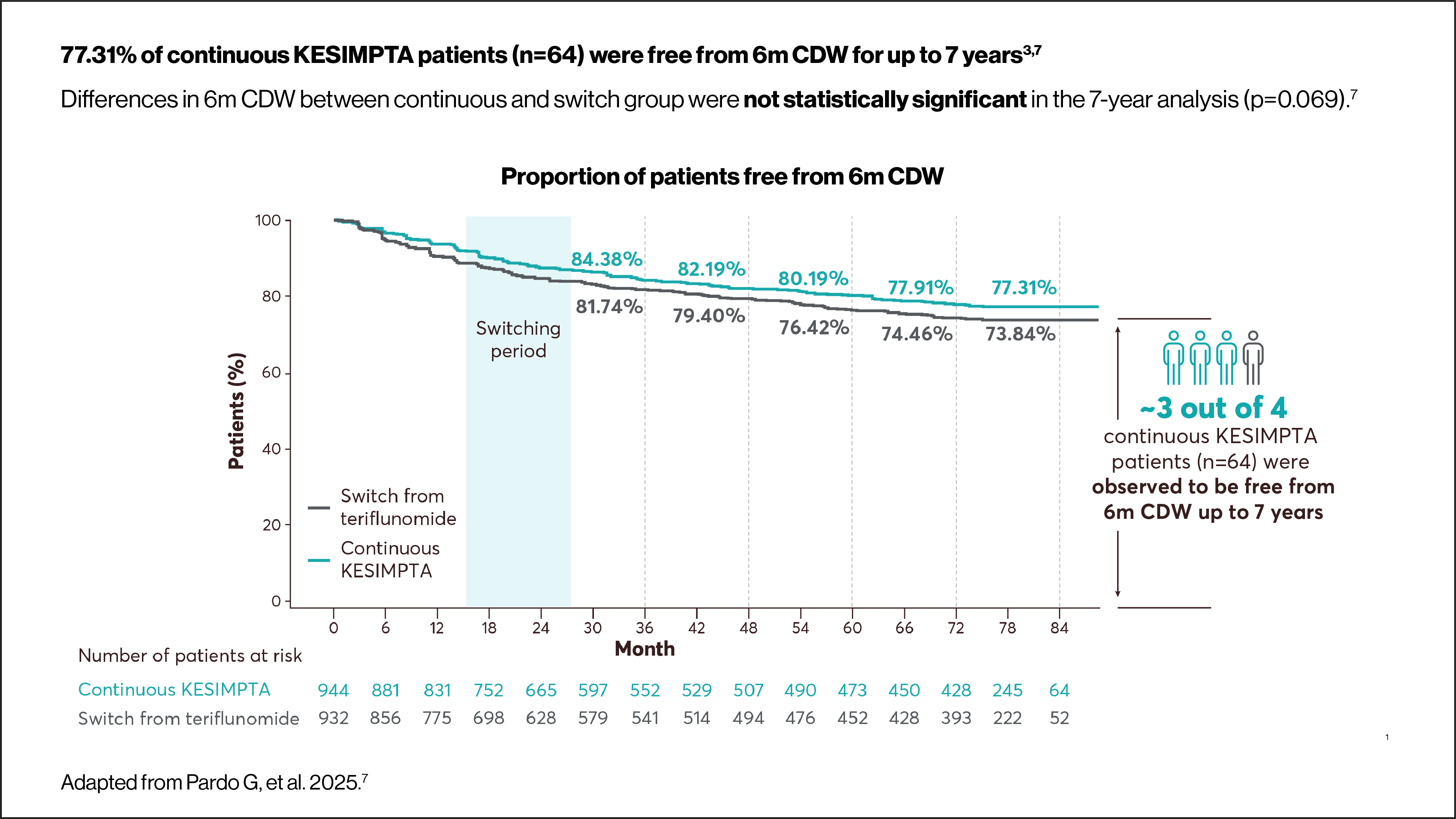
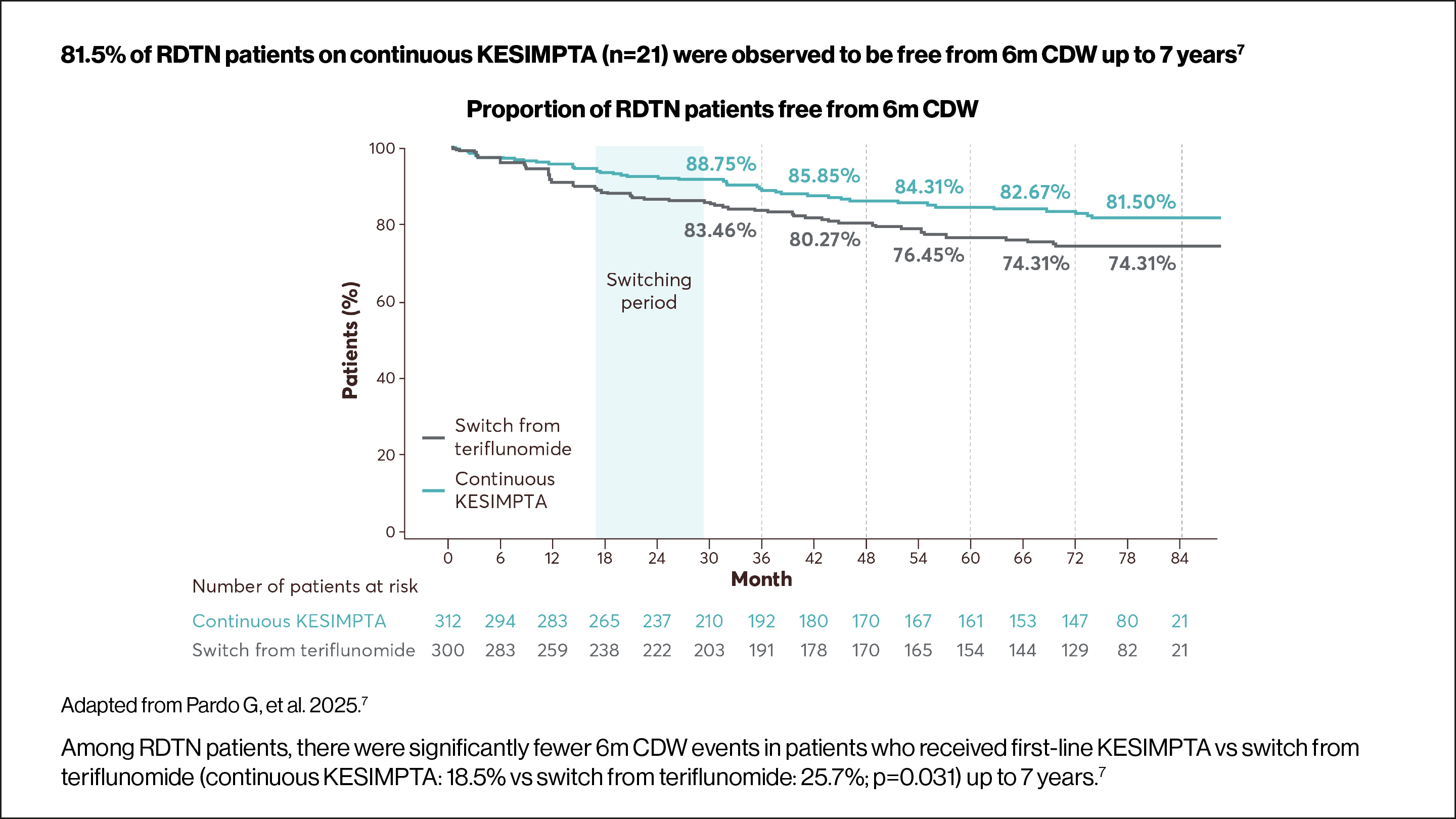
6m CDW was defined as an increase from baseline in EDSS score (≥1.5, ≥1 or ≥0.5 points for patients with a baseline EDSS score of 0, 1–5 or ≥5.5, respectively) sustained for ≥6 months. Time to first event rates were assessed using Kaplan–Meier estimates. For core+extension, p values were obtained using the log-rank test.7
Use the arrows below to navigate to the RDTN subgroup data.

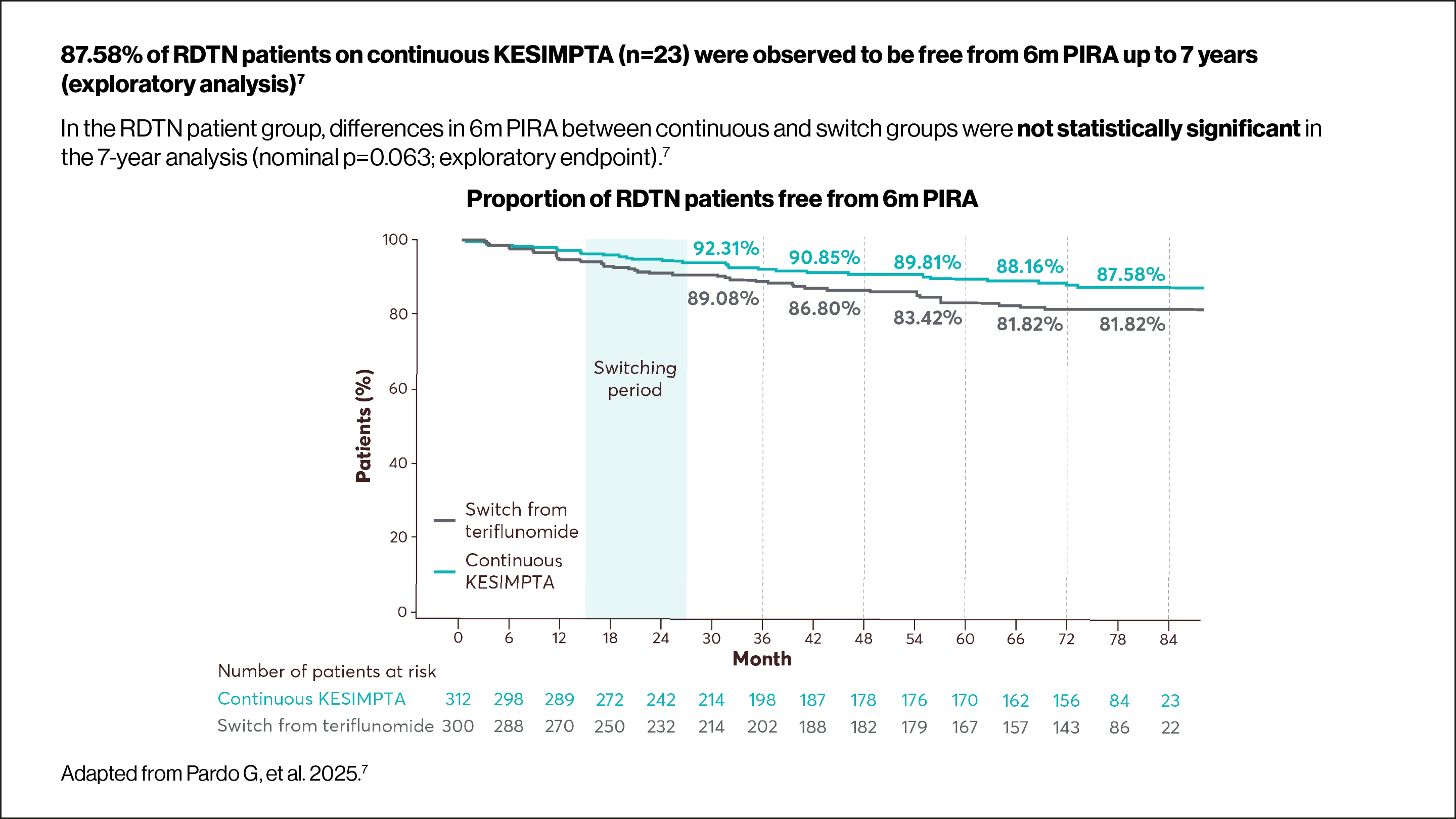
Data from exploratory analyses are descriptive, and no confirmatory clinical conclusions can be drawn.
6m PIRA was defined as a 6m CDW event with either no prior relapse or an onset more than 90 days after the start date of the last investigator-reported relapse. Additionally, to qualify as a PIRA event, no relapse must have occurred within 30 days after confirmation of EDSS worsening.7
Adapted from Novartis Data on File. 2025.9
% BVC estimates are obtained from a mixed model for repeated measures.9
§Statistically significant between-group comparison at each year for % BVC. All p values are nominal p values.9
Group | Core (%) | Extension (%) |
Continuous KESIMPTA | −0.34 | −0.27 |
Teriflunomide–KESIMPTA | −0.42 | −0.28 |
Between-group comparison | p=0.115 | p=0.666 |
Serum NfL levels during the overall period10
Adapted from Alvarez E, et al. 2023.10
*ARR was defined as the number of confirmed MS relapses per year, according to prespecified criteria. Confirmed relapses are those accompanied by a clinically relevant change in expanded disability status scale (EDSS).2
†ARRs are obtained from fitting a piecewise negative binomial model for the time period core phase and extension phase with log-link, adjusted for treatment and region as factors and number of relapses in previous year, baseline EDSS, baseline number of Gd+ lesions, and the participant’s age at baseline as covariates. The natural log of the time-in-study (in years) by period is used as offset to ARR in each period. Baseline variables are from the core study baseline. All p values are nominal p values. RDTN was defined as ≤3 years from diagnosis.6
‡Estimated from fitting a piecewise negative binomial model for the time period core phase and extension phase with log-link, adjusted for treatment as factor and baseline number of T1 Gd+ lesions and participant’s age at baseline as covariates. The natural log of the number of scans with evaluable Gd+ lesion counts by period is used as offset to obtain the lesion rate per scan in each period. Baseline variables are from the core study baseline. All p values are nominal p values.6
§Statistically significant between-group comparison at each year for % BVC. All p values are nominal p values.9
6m, 6-month; ABVC, annual rate of brain volume change; ARR, annualised relapse rate; BVC, brain volume change; CDI, confirmed disability improvement; CDW, confirmed disability worsening; CI, confidence interval; EDSS, expanded disability status scale; Gd+, gadolinium-enhancing; HR, hazard ratio; MRI, magnetic resonance imaging; MS, multiple sclerosis; NEDA-3, no evidence of disease activity-3; NfL, neurofilament light chain; OR, odds ratio; PIRA, progression independent of relapse activity; RDTN, recently-diagnosed treatment-naïve; RMS, relapsing forms of multiple sclerosis; RR, rate ratio; SmPC, summary of product characteristics; sNfL, serum neurofilament light chain.
References
KESIMPTA (ofatumumab) Summary of Product Characteristics.
Wiendl H, et al. Poster P9.010. American Academy of Neurology Annual Meeting. 13–18 April 2024, Denver, CO, US.
Hauser SL, et al. New Engl J Med 2020;383(6):546–557.
Hauser SL, et al. Mult Scler J 2022;28(10):1576–1590.
Hauser SL, et al. Poster P804. European Committee for Treatment and Research in Multiple Sclerosis Annual Congress. 24–26 September 2025, Barcelona, Spain.
Bittner S, et al. Poster P805. European Committee for Treatment and Research in Multiple clerosis Annual Congress. 24–26 September 2025, Barcelona, Spain.
Pardo G, et al. Poster P7.016. American Academy of Neurology Annual Meeting. 5–9 April 2025, San Diego, CA, US.
Cohen JA, et al. P009. American Academy of Neurology Annual Meeting. 22–27 April 2023, Boston, MA, US.
Novartis Data on File. OFA030. January 2025.
Alvarez E, et al. P6013. American Academy of Neurology Annual Meeting. 22–27 April 2023, Boston, MA, US.
UK | December 2025 | 443383-2
Adverse events should be reported. Reporting forms and information can be found at www.mhra.gov.uk/yellowcard. Adverse events should also be reported to Novartis online through the pharmacovigilance intake (PVI) tool at www.novartis.com/report, or alternatively email [email protected] or call 01276 698370.
*Other relevant decision makers particularly includes those with an NHS role who could influence in any way the administration, consumption, prescription, purchase, recommendation, sale, supply or use of any medicine but who are not health professionals.
This website is designed to be viewed on desktop.
 logo.")
 logo.")


 In Core Phase ASCLEPIOS I/II KESIMPTA showed a 54.2% relative reduction rate vs teriflunomide. ii) OPEN LABEL ASCLEPIOS EXTENSION: Continuous Kesimpta showed a 72.5% and a 54.8% relative reduction rate vs. teriflunomide-KESIMPTA.")
 and 93.8% on teriflunomide-KESIMPTA (n=584) were observed to have NEDA-3. i) Icon of abb calendar representing 6-month confirmed disability worsening (CDW). ii) Icon of a continuous flow chart representing confirmed relapse. iii) Icon of a hand representing Gd+ T1 lesions AND new or enlarging T2 lesions.")
.")
.")
 CORE PHASE ASCLEPIOS I/II: KESIMPTA showed a 96% relative reduction rate in Gd+ T1 lesions vs. teriflunomide. ii) OPEN LABEL ASCLEPIOS EXTENSION: KESIMPTA showed a 98% and a 57% relative reduction rate in Gd+ T1 lesions vs. teriflunomide.")
 CORE PHASE ASCLEPIOS I/II: KESIMPTA showed an 84% relative reduction rate in T2 lesions vs. teriflunomide. ii) OPEN LABEL ASCLEPIOS EXTENSION: KESIMPTA showed a 92% and a 89% relative reduction rate in T2 lesions vs. teriflunomide.")






