Resources
Explore our range of resources which have been designed to support you in your management of patients.
This page is intended for UK healthcare professionals and other relevant decision makers only. If you are a member of the public, please click here.
This portal is funded and owned by Novartis Pharmaceuticals UK Ltd and includes content approved by Novartis.
Adverse events reporting information can be found in the footer of this page.

JAKAVI® (ruxolitinib) is indicated for the treatment of disease-related splenomegaly or symptoms in adult patients with primary myelofibrosis (also known as chronic idiopathic myelofibrosis), post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis. JAKAVI® is also indicated for adult patients with polycythaemia vera who are resistant to or intolerant of hydroxyurea.1
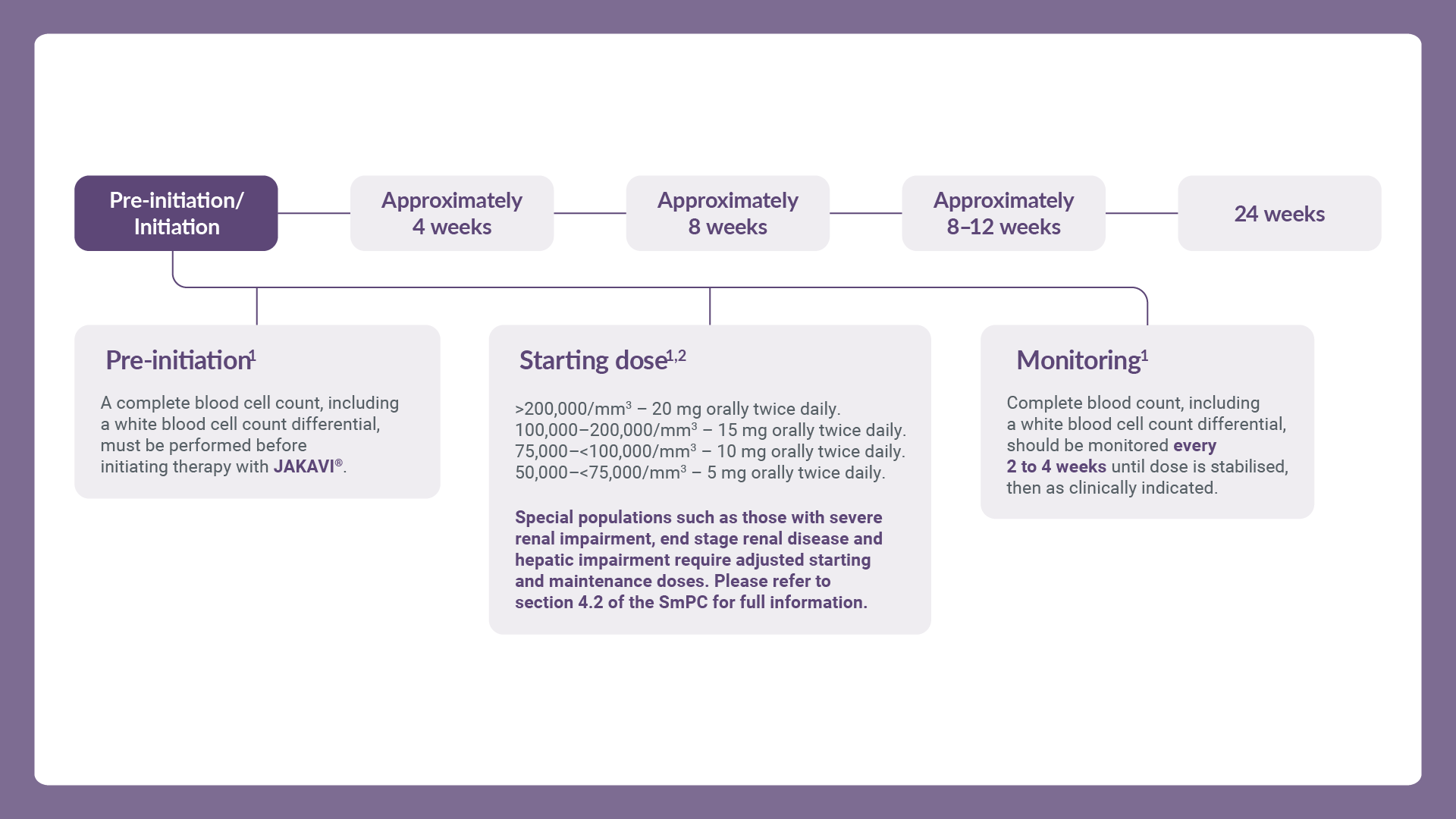
The recommended starting dose for JAKAVI® in myelofibrosis is based on a patient’s platelet count at treatment initiation.1
5 mg twice daily for patients with a platelet count between 50,000/mm3 and <75,000/mm3
10 mg twice daily for patients with a platelet count between 75,000/mm3 and <100,000/mm3
15 mg twice daily for patients with a platelet count between 100,000/mm3 and 200,000/mm3
20 mg twice daily for patients with a platelet count of >200,000/mm3
The recommended starting dose of JAKAVI® in polycythaemia vera (PV) is 10 mg given orally twice daily.1 For more information about JAKAVI® in PV, click here.
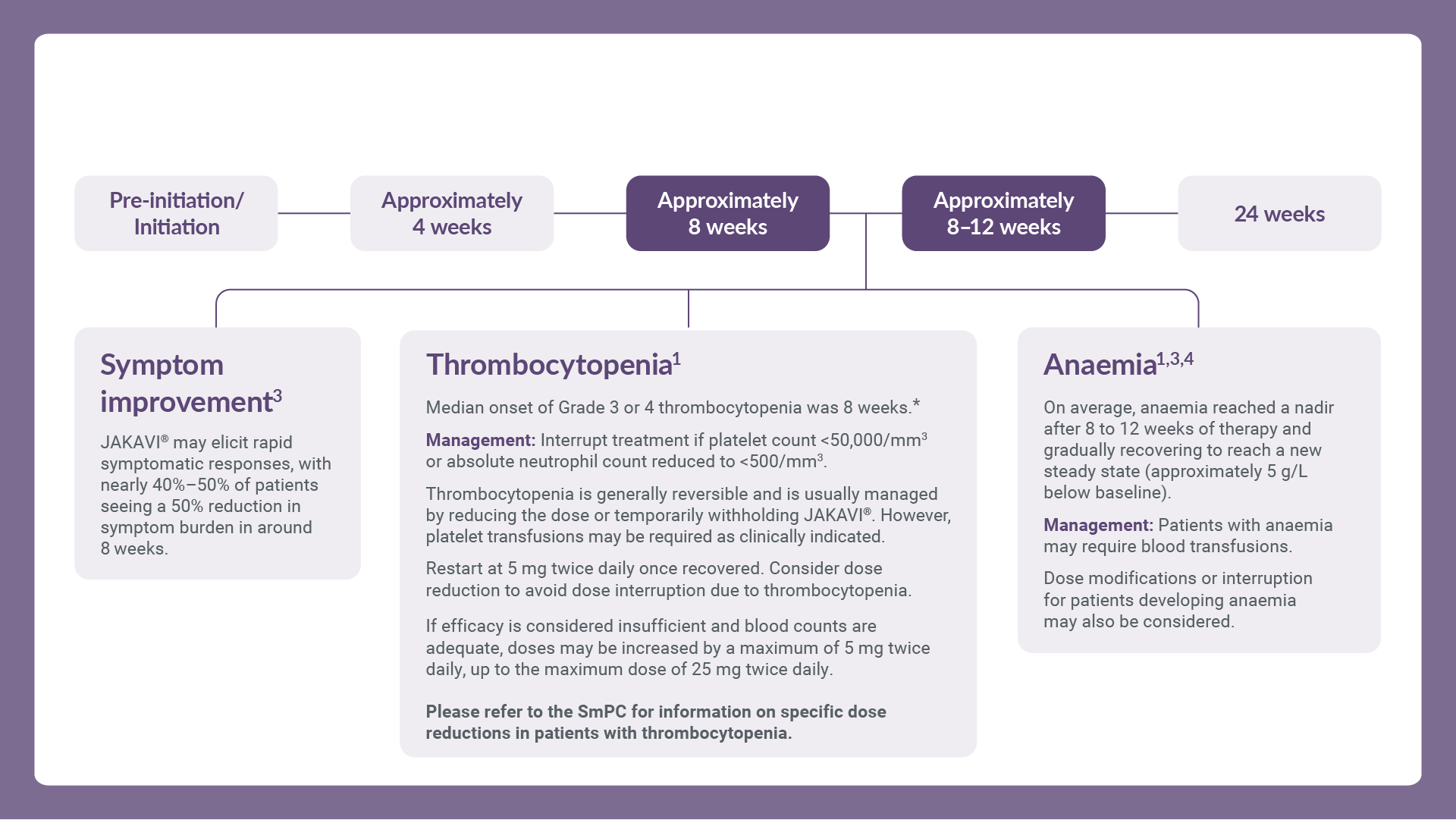
Treatment should be discontinued for platelet counts less than 50,000/mm3 or absolute neutrophil counts less than 500/mm3.1
After recovery of blood counts above these levels, dosing may be re-started at 5 mg twice daily and gradually increased based on careful monitoring of complete blood cell count, including a white blood cell count differential.1
JAKAVI® treatment should only be initiated by a physician experienced in the administration of anti-cancer medicinal products. A complete blood cell count, including a white blood cell count differential, must be performed before initiating therapy with JAKAVI®.
For further information, please refer to the SmPC.
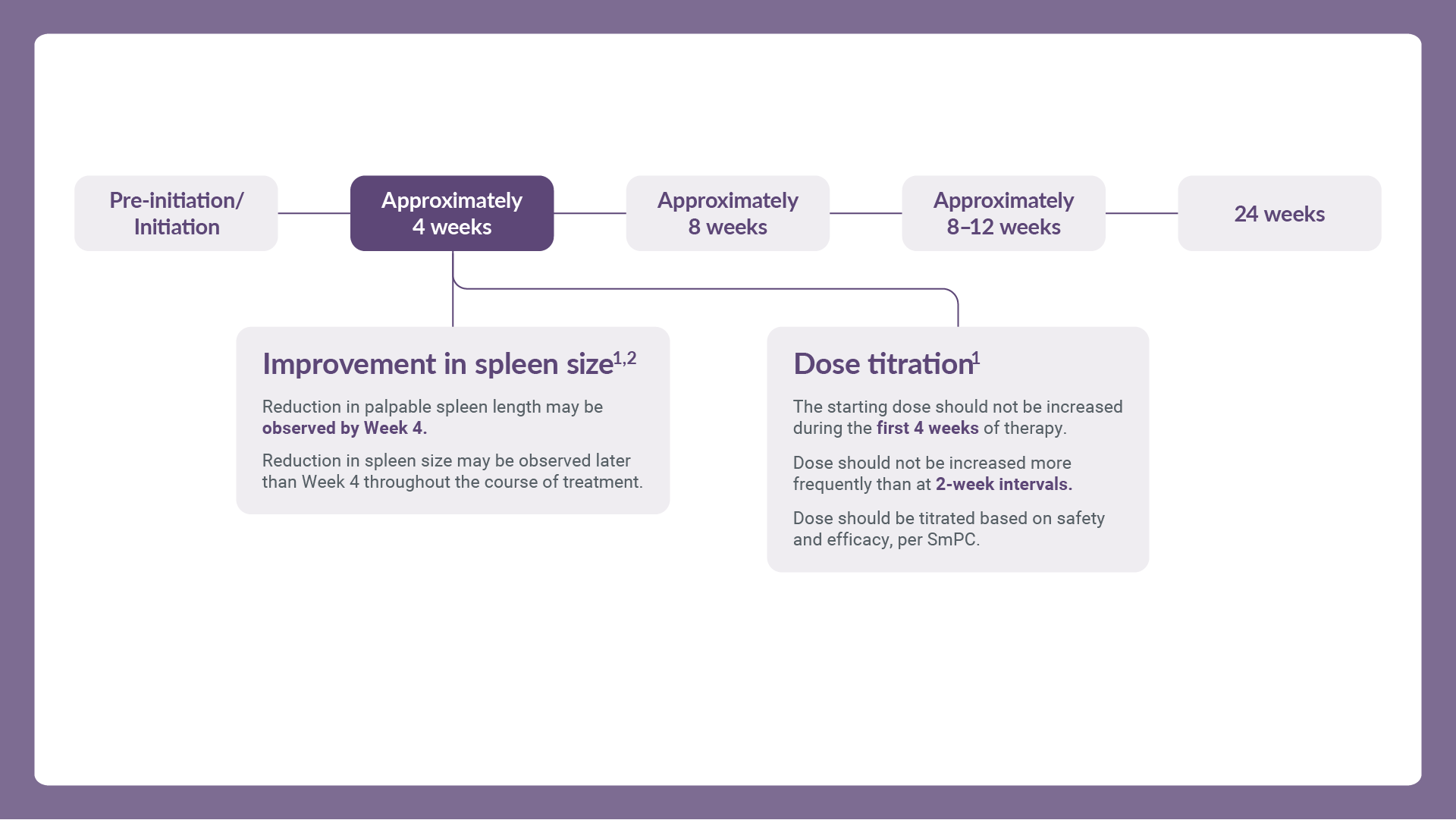

Treatment may continue as long as benefit–risk balance remains positive. After interruption or discontinuation of JAKAVI® dosing, symptoms of MF may return over a period of approximately 1 week. Unless abrupt discontinuation is required, gradual tapering of the dose may be considered, although the utility of tapering is unproven. Refer to the full SmPC for further management guidance.
Proactively monitor all patients’ MF-related symptoms to treat them as early as possible in those who are eligible for JAKAVI® and adjust dose as needed based on efficacy and safety profile.3–6
COMFORT-II 5-year follow-up study: mean haemoglobin levels over time7
Adapted from Harrison C, et al. 2015.7
For JAKAVI® patients who develop treatment-related anaemia:
If EPO <500 IU, consider adding:
If EPO >500 IU,§ consider adding:
No specific dose adjustment is needed in patients with mild or moderate renal impairment.
In patients with severe renal impairment (creatinine clearance less than 30 ml/min), the recommended starting dose based on platelet count for MF patients should be reduced by approximately 50% to be administered twice daily.
There are limited data to determine the best dosing options for patients with end-stage renal disease (ESRD) on haemodialysis. Pharmacokinetic/pharmacodynamic simulations based on available data in this population suggest that the starting dose for MF patients with ESRD on haemodialysis is a single dose of 15–20 mg or two doses of 10 mg given 12 hours apart, to be administered post-dialysis and only on the day of haemodialysis. A single dose of 15 mg is recommended for MF patients with platelet count between 100,000/mm3 and 200,000/mm3. A single dose of 20 mg or two doses of 10 mg given 12 hours apart is recommended for MF patients with platelet count of >200,000/mm3. Subsequent doses (single administration or two doses of 10 mg given 12 hours apart) should be administered only on haemodialysis days following each dialysis session.
In MF patients with any hepatic impairment, the recommended starting dose based on platelet count should be reduced by approximately 50% to be administered twice daily. Subsequent doses should be adjusted based on careful monitoring of safety and efficacy.
Patients diagnosed with hepatic impairment while receiving JAKAVI® should have complete blood counts, including a white blood cell count differential, monitored at least every one to two weeks for the first 6 weeks after initiation of therapy with JAKAVI® and as clinically indicated thereafter once their liver function and blood counts have been stabilised. JAKAVI® dose can be titrated to reduce the risk of cytopenia.1
No additional dose adjustments are recommended for elderly patients.
The safety and efficacy of JAKAVI® in children and adolescents aged up to 18 years with MF and PV have not been established. No data are available (see Section 5.1 of the SmPC).
If JAKAVI® is to be co-administered with strong CYP3A4 inhibitors in MF and PV patients or dual inhibitors of CYP3A4 and CYP2C9 enzymes (e.g., fluconazole), the unit dose of JAKAVI® should be reduced by approximately 50%, to be administered twice daily (for monitoring frequency, see sections 4.2 and 4.5 of the SmPC).
The concomitant use of cytoreductive therapies with JAKAVI® was associated with manageable cytopenias (see section 4.2 of the SmPC for dose modifications during cytopenias). Patients should be closely monitored (e.g. twice weekly) for cytopenias, and dose titrated based on safety and efficacy.
Avoid the concomitant use of JAKAVI® with fluconazole doses greater than 200 mg daily.
Please refer to the SmPC for full dosing information before prescribing.
The most frequently reported adverse drug reactions were thrombocytopenia and anaemia. Haematological adverse drug reactions (any Common Terminology Criteria for Adverse Events [CTCAE] grade) included anaemia (83.8%), thrombocytopenia (80.5%) and neutropenia (20.8%). Anaemia, thrombocytopenia and neutropenia are dose-related effects.1
*Grade 3 or 4 thrombocytopenia is defined as absolute platelet counts below 50,000/mm3.1
†In the absence of non-MF-related contributing factors.
‡BSH recommended starting dose: 20,000–40,000 units per week of recombinant EPO or darbepoetin 150 μg/week (or 500 μg/3 weeks) with dose escalation to 60,000 units or 300 μg per week, respectively, if required after 6–8 weeks. In the absence of beneficial response despite dose optimisation, ESAs should be discontinued.3
§Or the patient is refractory to ESAs.9
BAT, best available therapy; BSH, British Society for Haematology; CTCAE, Common Terminology Criteria for Adverse Events; CYP3A4, cytochrome P450 3A4; EPO, erythropoietin; ESA, erythropoiesis-stimulating agent; ESRD, end-stage renal disease; Hb, haemoglobin; IMiD, immunomodulatory drug; MF, myelofibrosis; PV, polycythaemia vera; SmPC, summary of product characteristics.
References
JAKAVI® (ruxolitinib) Summary of Product Characteristics.
Harrison C, et al. N Engl J Med 2012;366:787–798.
McLornan DP, et al. Br J Haematol 2024;204:136–150.
National Institute for Health and Care Excellence. Ruxolitinib for treating disease-related splenomegaly or symptoms in adults with myelofibrosis. TA386, 2016. Available at: https://www.nice.org.uk/guidance/ta386 [Accessed July 2025].
Mesa R, et al. Blood 2012;120:1727.
Scottish Medicines Consortium. available at: https://www.scottishmedicines.org.uk/medicines-advice/ruxolitinib-jakavi-fullsubmission-86713 [Accessed July 2025].
Harrison C, et al. ASH 2015, 5–8 December, Orlando, Florida, USA. Abstract 59.
Verstovsek S, Ann Hematol 2023;102(4):689–698.
Koschmieder S, et al. Leukemia 2024;38(8):1831–1838.
UK | September 2025 | FA-11210096-1
Adverse events should be reported. Reporting forms and information can be found at www.mhra.gov.uk/yellowcard. Adverse events should also be reported to Novartis online through the pharmacovigilance intake (PVI) tool at www.novartis.com/report, or alternatively email [email protected] or call 01276 698370.
*Other relevant decision makers particularly includes those with an NHS role who could influence in any way the administration, consumption, prescription, purchase, recommendation, sale, supply or use of any medicine but who are not health professionals.
This website is designed to be viewed on desktop.
 logo")
 logo")






